The MRPL3 Polyclonal Antibody (PACO06654) is a valuable tool for researchers studying the mitochondrial ribosomal protein L3 (MRPL3). This antibody is produced in rabbits and has a high reactivity with human samples, making it ideal for use in Western blot applications. By binding to the MRPL3 protein, this antibody enables the detection and analysis of MRPL3 in various cell types.MRPL3 is a key component of the mitochondrial ribosome and plays a crucial role in protein synthesis within the mitochondria. Research has shown that MRPL3 mutations can lead to mitochondrial dysfunction, which is implicated in a variety of diseases including cancer, neurodegenerative disorders, and metabolic conditions.
Understanding the function and regulation of MRPL3 is essential for unraveling the molecular mechanisms underlying these diseases and developing targeted therapies.By using the MRPL3 Polyclonal Antibody, researchers can gain insights into the role of MRPL3 in mitochondrial biology and disease pathology, ultimately advancing our understanding of mitochondrial dysfunction and its implications for human health.
Antibody Name:
MRPL3 Antibody (PACO06654)
Antibody SKU:
PACO06654
Size:
50ug
Host Species:
Rabbit
Tested Applications:
ELISA, WB
Recommended Dilutions:
ELISA:1:5000, WB:1:500-1:2000
Species Reactivity:
Human
Immunogen:
Synthesized peptide derived from the Internal region of human MRP-L3.
Form:
Liquid
Storage Buffer:
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Purification Method:
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Clonality:
Polyclonal
Isotype:
IgG
Conjugate:
Non-conjugated
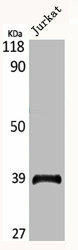
Western Blot analysis of Jurkat cells using MRP-L3 Polyclonal Antibody.
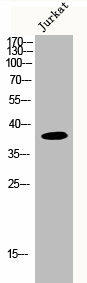
Western Blot analysis of Jurkat cells using MRP-L3 Polyclonal Antibody.
Synonyms:
MRPL3; MRL3; RPML3; 39S ribosomal protein L3; mitochondrial; L3mt; MRP-L3
UniProt Protein Function:
MRPL3: Defects in MRPL3 are the cause of combined oxidative phosphorylation deficiency type 9 (COXPD9). A mitochondrial disease characterized by failure to thrive, poor feeding, hypertrophic cardiomyopathy, hepatomegaly, and psychomotor retardation. Death in infancy has been observed in some cases. Belongs to the ribosomal protein L3P family.Protein type: Mitochondrial; Ribosomal; Translation; RNA-bindingChromosomal Location of Human Ortholog: 3q21-q23Cellular Component: mitochondrial inner membrane; mitochondrial large ribosomal subunitMolecular Function: structural constituent of ribosome; RNA bindingBiological Process: mitochondrial translation; translation; organelle organization and biogenesisDisease: Combined Oxidative Phosphorylation Deficiency 9
UniProt Protein Details:
NCBI Summary:
Mammalian mitochondrial ribosomal proteins are encoded by nuclear genes and help in protein synthesis within the mitochondrion. Mitochondrial ribosomes (mitoribosomes) consist of a small 28S subunit and a large 39S subunit. They have an estimated 75% protein to rRNA composition compared to prokaryotic ribosomes, where this ratio is reversed. Another difference between mammalian mitoribosomes and prokaryotic ribosomes is that the latter contain a 5S rRNA. Among different species, the proteins comprising the mitoribosome differ greatly in sequence, and sometimes in biochemical properties, which prevents easy recognition by sequence homology. This gene encodes a 39S subunit protein that belongs to the L3P ribosomal protein family. A pseudogene corresponding to this gene is found on chromosome 13q. [provided by RefSeq, Jul 2008]












")


