The PKHD1 Polyclonal Antibody (PAC050362) is a valuable tool for researchers studying the protein PKHD1, which is involved in various cellular processes and has been linked to polycystic kidney diseases. Raised in rabbits, this antibody is highly specific to human samples and has been validated for use in Western blot applications. By binding to the PKHD1 protein, this antibody allows for the detection and analysis of PKHD1 expression in a variety of cell types, making it ideal for studies in kidney disease research and cell biology.PKHD1 is a large and complex protein that plays a crucial role in kidney development and function.
Mutations in the PKHD1 gene have been associated with various forms of polycystic kidney diseases, which are characterized by the formation of fluid-filled cysts in the kidneys. Understanding the function and regulation of PKHD1 is essential for uncovering the mechanisms underlying these diseases and developing potential treatments. The PKHD1 Polyclonal Antibody is a valuable tool for researchers seeking to investigate the role of PKHD1 in health and disease.
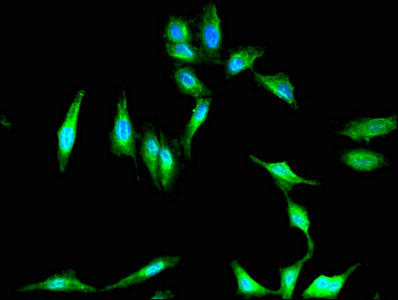
Immunofluorescent analysis of Hela cells using PACO50362 at dilution of 1:100 and Alexa Fluor 488-congugated AffiniPure Goat Anti-Rabbit IgG(H+L).
Western Blot. Positive WB detected in Recombinant protein. All lanes: PKHD1 antibody at 3µg/ml. Secondary. Goat polyclonal to rabbit IgG at 1/50000 dilution. predicted band size: 55 kDa. observed band size: 55 kDa.
Background:
May be required for correct bipolar cell division through the regulation of centrosome duplication and mitotic spindle assembly. May be a receptor protein that acts in collecting-duct and biliary differentiation.
PKHD1: May be required for correct bipolar cell division through the regulation of centrosome duplication and mitotic spindle assembly. May be a receptor protein that acts in collecting-duct and biliary differentiation. Defects in PKHD1 are the cause of polycystic kidney disease autosomal recessive (ARPKD). ARPKD is a severe form of polycystic kidney disease affecting the kidneys and the hepatic biliary tract. The clinical spectrum is widely variable, with most cases presenting during infancy. The fetal phenotypic features classically include enlarged and echogenic kidneys, as well as oligohydramnios secondary to a poor urine output. Up to 50% of the affected neonates die shortly after birth, as a result of severe pulmonary hypoplasia and secondary respiratory insufficiency. In the subset that survives the perinatal period, morbidity and mortality are mainly related to severe systemic hypertension, renal insufficiency, and portal hypertension due to portal-tract fibrosis. 2 isoforms of the human protein are produced by alternative splicing.Protein type: Cell surface; Membrane protein, integralChromosomal Location of Human Ortholog: 6p12.2Cellular Component: anchored to external side of plasma membrane; apical plasma membrane; centrosome; cilium; cytoplasm; perinuclear region of cytoplasmMolecular Function: protein bindingBiological Process: cell-cell adhesion; cellular calcium ion homeostasis; inhibition of NF-kappaB transcription factor; kidney development; negative regulation of apoptosis; negative regulation of cell motility; negative regulation of protein kinase B signaling cascade; positive regulation of cell proliferation; regulation of TOR signaling pathwayDisease: Polycystic Kidney Disease, Autosomal Recessive
UniProt Protein Details:
NCBI Summary:
The protein encoded by this gene is predicted to have a single transmembrane (TM)-spanning domain and multiple copies of an immunoglobulin-like plexin-transcription-factor domain. Alternative splicing results in two transcript variants encoding different isoforms. Other alternatively spliced transcripts have been described, but the full length sequences have not been determined. Several of these transcripts are predicted to encode truncated products which lack the TM and may be secreted. Mutations in this gene cause autosomal recessive polycystic kidney disease, also known as polycystic kidney and hepatic disease-1. [provided by RefSeq, Jul 2008]






.")
.")

.")


.")