The MPDU1 Antibody (PAC028306) is a vital tool for researchers studying the MPDU1 protein, an essential enzyme involved in the biosynthesis of dolichol-linked oligosaccharides. This monoclonal antibody, produced using hybridoma technology, exhibits high specificity and sensitivity towards MPDU1 in human samples.MPDU1 is a key player in the process of N-linked glycosylation, a crucial post-translational modification that influences protein folding, stability, and function. Dysregulation of this pathway has been implicated in various diseases, including congenital disorders and cancer. By targeting MPDU1 with this antibody, researchers can investigate its role in glycosylation pathways and its potential as a therapeutic target.
This antibody is validated for use in a variety of applications, including Western blotting, immunofluorescence, and immunohistochemistry, providing researchers with versatile options for studying MPDU1 expression and localization in different cell types and tissues. With its high specificity and reliability, the MPDU1 Antibody (PAC028306) is a valuable tool for advancing research in the fields of glycobiology, protein processing, and disease pathology.
Immunohistochemistry of paraffin-embedded human brain tissue using PACO28306 at dilution of 1:100.
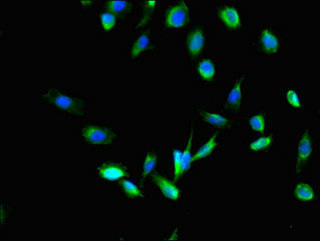
Immunofluorescent analysis of U251 cells using PACO28306 at dilution of 1:100 and Alexa Fluor 488-congugated AffiniPure Goat Anti-Rabbit IgG(H+L).
Background:
Required for normal utilization of mannose-dolichol phosphate (Dol-P-Man) in the synthesis of N-linked and O-linked oligosaccharides and GPI anchors.
Synonyms:
Mannose-P-dolichol utilization defect 1 protein (Suppressor of Lec15 and Lec35 glycosylation mutation homolog) (SL15), MPDU1
UniProt Protein Function:
MPDU1: Required for normal utilization of mannose-dolichol phosphate (Dol-P-Man) in the synthesis of N-linked and O-linked oligosaccharides and GPI anchors. Defects in MPDU1 are the cause of congenital disorder of glycosylation type 1F (CDG1F). CDGs are a family of severe inherited diseases caused by a defect in protein N- glycosylation. They are characterized by under-glycosylated serum proteins. These multisystem disorders present with a wide variety of clinical features, such as disorders of the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. Belongs to the MPDU1 (TC 2.A.43.3) family.Protein type: Membrane protein, multi-pass; Membrane protein, integralChromosomal Location of Human Ortholog: 17p13.1-p12Cellular Component: endoplasmic reticulum; endoplasmic reticulum membrane; integral to membrane; membrane; mitochondrionMolecular Function: protein bindingBiological Process: dolichol-linked oligosaccharide biosynthetic process; oligosaccharide biosynthetic process; protein folding; transportDisease: Congenital Disorder Of Glycosylation, Type If
UniProt Protein Details:
NCBI Summary:
This gene encodes an endoplasmic reticulum membrane protein that is required for utilization of the mannose donor mannose-P-dolichol in the synthesis of lipid-linked oligosaccharides and glycosylphosphatidylinositols. Mutations in this gene result in congenital disorder of glycosylation type If. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Dec 2008]




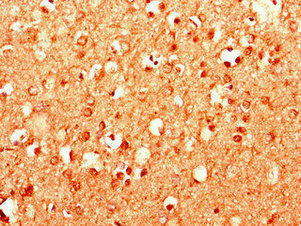
. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.")
. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.")

.")


.")

.")