The KCNQ4 Polyclonal Antibody (PAC019895) is an essential tool for researchers studying the KCNQ4 protein, a voltage-gated potassium channel involved in auditory function. This antibody, produced in rabbits, demonstrates high specificity and sensitivity towards human samples, making it ideal for use in Western blot applications. By targeting the KCNQ4 protein, researchers can easily detect and analyze its expression in various cell types, facilitating investigations into the role of KCNQ4 in auditory physiology and pathology.
KCNQ4, also known as Kv7.4, is crucial for maintaining the electrical properties of hair cells in the cochlea and regulating auditory transduction. Dysregulation of KCNQ4 has been implicated in hereditary hearing loss and other auditory disorders, making it a promising target for therapeutic interventions. By studying the function of KCNQ4, researchers can gain insights into the mechanisms underlying hearing loss and potentially develop novel treatment strategies to protect and restore auditory function.
The image on the left is immunohistochemistry of paraffin-embedded Human brain tissue using PACO19895(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: x200).
Gel: 6%SDS-PAGE, Lysate: 40 μg, Lane 1-2: Human fetal brain tissue, mouse brain tissue, Primary antibody: PACO19895(KCNQ4 Antibody) at dilution 1/200, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 7 minutes.
The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using PACO19895(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: x200).
Background:
The protein encoded by this gene forms a potassium channel that is thought to play a critical role in the regulation of neuronal excitability, particularly in sensory cells of the cochlea. The current generated by this channel is inhibited by M1 muscarinic acetylcholine receptors and activated by retigabine, a novel anti-convulsant drug. The encoded protein can form a homomultimeric potassium channel or possibly a heteromultimeric channel in association with the protein encoded by the KCNQ3 gene. Defects in this gene are a cause of nonsyndromic sensorineural deafness type 2 (DFNA2), an autosomal dominant form of progressive hearing loss. Two transcript variants encoding different isoforms have been found for this gene.
Synonyms:
potassium voltage-gated channel, KQT-like subfamily, member 4







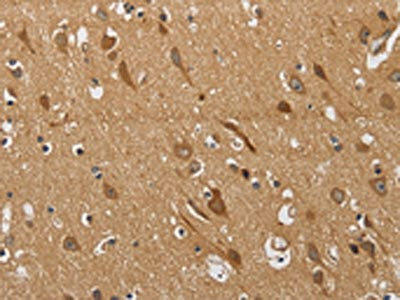
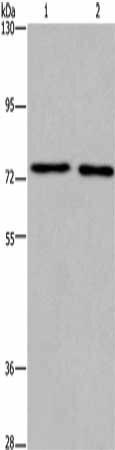
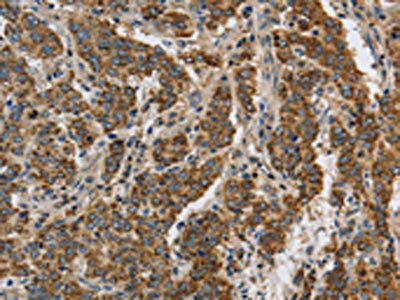





. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.")
. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.")

.")