The IMPAD1 Polyclonal Antibody (PACO19838) is a valuable tool for researchers studying the IMPAD1 protein, a key player in lipid metabolism and cellular function. This antibody, produced in rabbits, is highly specific for human samples and has been validated for use in Western blot applications. By targeting the IMPAD1 protein, researchers can gain valuable insights into its role in lipid processing and its potential implications for metabolic disorders and other related conditions.IMPAD1, also known as inositol monophosphatase domain containing 1, is involved in various cellular processes, including lipid biosynthesis and metabolism.
Dysregulation of IMPAD1 has been implicated in metabolic disorders such as diabetes and obesity, making it a promising target for therapeutic interventions. By using the IMPAD1 Polyclonal Antibody, researchers can further explore the mechanisms underlying IMPAD1's function and its potential as a therapeutic target for metabolic diseases.
Antibody Name:
IMPAD1 Antibody (PACO19838)
Antibody SKU:
PACO19838
Size:
50ul
Host Species:
Rabbit
Tested Applications:
ELISA, IHC
Recommended Dilutions:
ELISA:1:1000-1:2000, IHC:1:25-1:100
Species Reactivity:
Human, Mouse, Rat
Immunogen:
Synthetic peptide of human IMPAD1
Form:
Liquid
Storage Buffer:
-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
Purification Method:
Antigen affinity purification
Clonality:
Polyclonal
Isotype:
IgG
Conjugate:
Non-conjugated
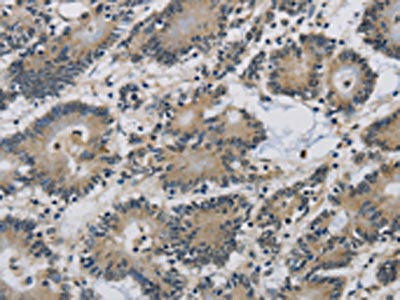
The image on the left is immunohistochemistry of paraffin-embedded Human colon cancer tissue using PACO19838(IMPAD1 Antibody) at dilution 1/20, on the right is treated with synthetic peptide. (Original magnification: x200).
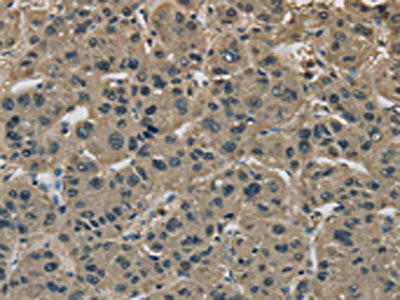
The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using PACO19838(IMPAD1 Antibody) at dilution 1/20, on the right is treated with synthetic peptide. (Original magnification: x200).
Background:
This gene encodes a member of the inositol monophosphatase family. The encoded protein is localized to the Golgi apparatus and catalyzes the hydrolysis of phosphoadenosine phosphate (PAP) to adenosine monophosphate (AMP). Mutations in this gene are a cause of GRAPP type chondrodysplasia with joint dislocations, and a pseudogene of this gene is located on the long arm of chromosome 1.
Synonyms:
inositol monophosphatase domain containing 1
UniProt Protein Function:
IMPAD1: May play a role in the formation of skeletal elements derived through endochondral ossification, possibly by clearing adenosine 3',5'-bisphosphate produced by Golgi sulfotransferases during glycosaminoglycan sulfation. Defects in IMPAD1 are the cause of chondrodysplasia with joint dislocations GPAPP type (CDP-GPAPP). A condition consisting of congenital joint dislocations, chondrodysplasia with short stature, micrognathia and cleft palate, and a distinctive face. Belongs to the inositol monophosphatase family.Protein type: Membrane protein, integral; EC 3.1.3.25; Phosphatase (non-protein); EC 3.1.3.7Chromosomal Location of Human Ortholog: 8q12.1Cellular Component: Golgi apparatus; membrane; integral to membraneMolecular Function: 3'-nucleotidase activity; metal ion binding; inositol-1(or 4)-monophosphatase activity; 3'(2'),5'-bisphosphate nucleotidase activityBiological Process: chondroitin sulfate metabolic process; dephosphorylation; phosphoinositide phosphorylation; inositol biosynthetic process; embryonic digit morphogenesis; chondrocyte development; endochondral ossification; post-embryonic developmentDisease: Chondrodysplasia With Joint Dislocations, Gpapp Type
UniProt Protein Details:
NCBI Summary:
This gene encodes a member of the inositol monophosphatase family. The encoded protein is localized to the Golgi apparatus and catalyzes the hydrolysis of phosphoadenosine phosphate (PAP) to adenosine monophosphate (AMP). Mutations in this gene are a cause of GRAPP type chondrodysplasia with joint dislocations, and a pseudogene of this gene is located on the long arm of chromosome 1. [provided by RefSeq, Dec 2011]







")


.")

