The IFT80 Antibody (PAC042022) is a highly specific tool for researchers studying the protein IFT80, which is involved in intraflagellar transport and ciliogenesis pathways. This polyclonal antibody, generated in rabbits, is optimized for detecting IFT80 in human and mouse samples through Western blot analysis. Its high specificity and sensitivity make it a valuable tool for investigating the role of IFT80 in various cellular processes, including ciliary assembly, maintenance, and signaling.IFT80 is a key component of the intraflagellar transport complex, essential for the formation and function of primary cilia.
Dysfunction in ciliary pathways has been linked to a range of human disorders known as ciliopathies, including developmental abnormalities, sensory impairments, and cilia-related diseases. By targeting IFT80 with this antibody, researchers can gain insights into the molecular mechanisms underlying cilia biogenesis and function, offering potential avenues for therapeutic interventions in ciliopathy-related conditions.
Antibody Name:
IFT80 Antibody (PACO42022)
Antibody SKU:
PACO42022
Size:
50ug
Host Species:
Rabbit
Tested Applications:
ELISA, IHC
Recommended Dilutions:
ELISA:1:2000-1:10000, IHC:1:20-1:200
Species Reactivity:
Human
Immunogen:
Recombinant Human Intraflagellar transport protein 80 homolog protein (1-268AA)
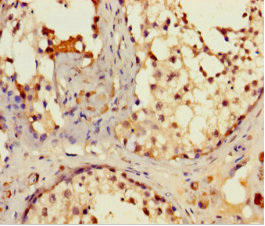
Immunohistochemistry of paraffin-embedded human testis tissue using PACO42022 at dilution of 1:100.
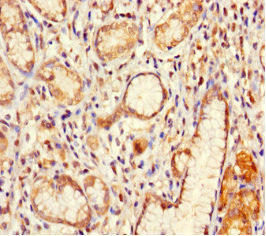
Immunohistochemistry of paraffin-embedded human gastric cancer using PACO42022 at dilution of 1:100.
Background:
Component of the intraflagellar transport (IFT) complex B, which is essential for the development and maintenance of motile and sensory cilia.
Synonyms:
Intraflagellar transport protein 80 homolog (WD repeat-containing protein 56), IFT80, KIAA1374 WDR56
UniProt Protein Function:
IFT80: Component of the intraflagellar transport (IFT) complex B, which is essential for the development and maintenance of motile and sensory cilia. Defects in IFT80 are the cause of asphyxiating thoracic dystrophy type 2 (ATD2). An autosomal recessive chondrodysplasia characterized by a severely constricted thoracic cage, short-limbed short stature, and polydactyly. It often leads to death in infancy because of respiratory insufficiency. Retinal degeneration, cystic renal disease and hepatic disease can be present in affected individuals who survive early childhood.Chromosomal Location of Human Ortholog: 3q25.33Cellular Component: centrosome; cilium; cytoplasmBiological Process: chondrocyte differentiation; cilium biogenesis; negative regulation of epithelial cell proliferation; osteoblast differentiation; positive regulation of smoothened signaling pathway; sensory cilium biogenesis; smoothened signaling pathwayDisease: Short-rib Thoracic Dysplasia 2 With Or Without Polydactyly
UniProt Protein Details:
NCBI Summary:
The protein encoded by this gene is part of the intraflagellar transport complex B and is necessary for the function of motile and sensory cilia. Defects in this gene are a cause of asphyxiating thoracic dystrophy 2 (ATD2). Three transcript variants encoding two different isoforms have been found for this gene.[provided by RefSeq, Jun 2010]





.")


.")

.")
.")