The EPM2A Antibody (PAC045806) offered by Assay Genie is a valuable tool for researchers studying the EPM2A protein, which is associated with a rare and severe form of epilepsy known as Lafora disease. This polyclonal antibody, produced in rabbits, has been rigorously validated for use in various research applications, including Western blotting.EPM2A is a critical enzyme involved in glycogen metabolism, and mutations in the EPM2A gene can lead to the accumulation of abnormal glycogen in the brain, causing neurological symptoms characteristic of Lafora disease. By targeting the EPM2A protein, researchers can gain insights into the molecular mechanisms underlying this devastating disorder and potentially identify new therapeutic targets for treatment.
With its high reactivity to human samples, the EPM2A Antibody is an essential tool for investigating the role of EPM2A in Lafora disease pathogenesis and exploring potential treatment strategies. Researchers in the fields of neurology, genetics, and molecular biology will find this antibody to be invaluable for their studies on this rare and debilitating neurological disorder.
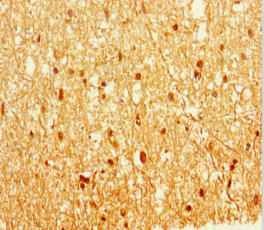
Immunohistochemistry of paraffin-embedded human brain tissue using PACO45806 at dilution of 1:100.
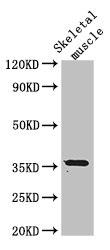
Western Blot. Positive WB detected in: Mouse skeletal muscle tissue. All lanes: EPM2A antibody at 3µg/ml. Secondary. Goat polyclonal to rabbit IgG at 1/50000 dilution. Predicted band size: 36 kDa. Observed band size: 36 kDa.
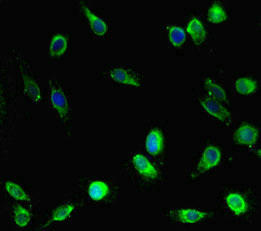
Immunofluorescent analysis of Hela cells using PACO45806 at dilution of 1:100 and Alexa Fluor 488-congugated AffiniPure Goat Anti-Rabbit IgG(H+L).
Background:
negative regulation of TOR signaling, positive regulation of macroautophagy
Synonyms:
Laforin, isoform 9, EPM2A
UniProt Protein Function:
laforin iso9: Dual specificity protein phosphatase. May be involved in the control of glycogen metabolism, particularly in monitoring for and preventing the formation of poorly branched glycogen molecules (polyglucosans). Acts as a scaffold protein to facilitate PPP1R3C/PTG ubiquitination by NHLRC1/malin. Forms a complex with NHLRC1/malin and HSP70 and this complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system (UPS). Isoform 2, an inactive phosphatase, could function as a dominant-negative regulator for the phosphatase activity of isoform 1. Defects in EPM2A are a cause of progressive myoclonic epilepsy type 2 (EPM2); also known as Lafora disease. EPM2 is an autosomal recessive and severe form of adolescent-onset progressive epilepsy. Typically, as seizures increase in frequency, cognitive function declines towards dementia, and affected individuals die usually within 10 years after onset. EPM2 occurs worldwide, but it is particularly common in the mediterranean countries of southern Europe and northern Africa, in southern India and in the Middle East. At the cellular level, it is characterized by accumulation of starch-like polyglucosans called Lafora bodies (LBs) that are most abundant in organs with the highest glucose metabolism: brain, heart, liver and skeletal muscle. Among other conditions involving polyglucosans, EPM2 is unique in that the inclusions are in neuronal dendrites but not axons and the forming polyglucosan fibrils are associated with the endoplasmic reticulum. Belongs to the protein-tyrosine phosphatase family. 9 isoforms of the human protein are produced by alternative splicing.Protein type: Protein phosphatase, dual-specificity; EC 3.1.3.16; EC 3.1.3.48; Motility/polarity/chemotaxisChromosomal Location of Human Ortholog: 6q24Biological Process: negative regulation of TOR signaling pathway; positive regulation of macroautophagy
UniProt Protein Details:
NCBI Summary:
This gene encodes a dual-specificity phosphatase that associates with polyribosomes. The encoded protein may be involved in the regulation of glycogen metabolism. Mutations in this gene have been associated with myoclonic epilepsy of Lafora. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2008]












.")
.")
. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit IgG labeled by HRP and visualized using 0.05% DAB.")
.")
