The CLCN7 Polyclonal Antibody (CAB6886) is a valuable tool for researchers studying CLCN7, a protein involved in bone resorption and lysosomal function. This antibody, produced in rabbits, exhibits high reactivity with human samples and has been validated for use in Western blot applications. By binding specifically to the CLCN7 protein, this antibody allows for precise detection and analysis in a variety of cell types, making it an essential reagent for studies in osteoporosis, lysosomal storage disorders, and other bone-related diseases.CLCN7, also known as chloride channel 7, plays a crucial role in maintaining bone homeostasis and acidification of lysosomes.
Dysregulation of CLCN7 function has been linked to various skeletal disorders, making it a promising target for therapeutic intervention. Understanding the mechanisms underlying CLCN7 activity is essential for developing treatments that target bone resorption and lysosomal dysfunction, with potential applications in treating osteoporosis and other bone disorders. In conclusion, the CLCN7 Polyclonal Antibody is an indispensable tool for researchers investigating the role of CLCN7 in bone physiology and disease. Its high specificity and reliability make it a valuable asset for studies aiming to uncover novel therapeutic approaches for bone-related disorders.
Product Name:
CLCN7 Rabbit Polyclonal Antibody
SKU:
CAB6886
Size:
20uL, 100uL
Isotype:
IgG
Host Species:
Rabbit
Reactivity:
Human,Mouse,Rat
Immunogen:
Recombinant fusion protein containing a sequence corresponding to amino acids 626-805 of human CLCN7 (NP_001278.1).
The product of this gene belongs to the CLC chloride channel family of proteins. Chloride channels play important roles in the plasma membrane and in intracellular organelles. This gene encodes chloride channel 7. Defects in this gene are the cause of osteopetrosis autosomal recessive type 4 (OPTB4), also called infantile malignant osteopetrosis type 2 as well as the cause of autosomal dominant osteopetrosis type 2 (OPTA2), also called autosomal dominant Albers-Schonberg disease or marble disease autosoml dominant. Osteopetrosis is a rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. OPTA2 is the most common form of osteopetrosis, occurring in adolescence or adulthood.
Purification Method:
Affinity purification
Gene ID:
1186
Storage Buffer:
Store at -20℃. Avoid freeze / thaw cycles.Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3.
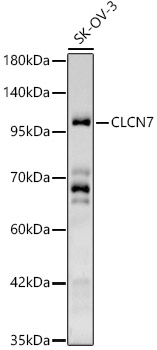
Western blot analysis of SK-OV-3, using CLCN7 Rabbit pAb (CAB6886) at 1:500 dilution.Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution.Lysates/proteins: 25ug per lane.Blocking buffer: 3% nonfat dry milk in TBST.Detection: ECL Basic Kit (AbGn00020).Exposure time: 180s.