The SMN1 Monoclonal Antibody (CAB9707) is a crucial tool for researchers studying spinal muscular atrophy (SMA), a genetic disorder characterized by the loss of motor neurons in the spinal cord. This antibody, produced in mice, is highly specific to the survival motor neuron (SMN) protein, particularly SMN1. Validated for use in various applications including immunohistochemistry and flow cytometry, it allows for the detection and analysis of SMN1 levels in different cell types.SMN1 is essential for the assembly of small nuclear ribonucleoproteins (snRNPs) which are involved in RNA splicing, making it a key player in gene expression regulation. Mutations in the SMN1 gene lead to decreased levels of SMN protein, resulting in motor neuron degeneration and subsequent muscle weakness in SMA patients.
Research into SMN1 function and regulation is crucial for developing targeted therapies for SMA and potentially other neurodegenerative disorders.Elevated levels of SMN1 have also been implicated in certain types of cancer, making this antibody a valuable tool for studying SMN1's role in tumorigenesis and potential therapeutic targeting. By understanding the mechanisms underlying SMN1 activity, researchers can make significant strides in both neurodegenerative disease and cancer research, ultimately leading to improved treatment options for patients.
Product Name:
SMN1 Rabbit Monoclonal Antibody
SKU:
CAB9707
Size:
20uL, 100uL
Isotype:
IgG
Host Species:
Rabbit
Reactivity:
Human,Rat
Immunogen:
Recombinant fusion protein containing a sequence corresponding to amino acids 103-200 of human SMN1 (Q16637).
Cajal body, Cytoplasm, Cytoplasmic ribonucleoprotein granule, Cytosol, Gemini of coiled bodies, Nuclear body, Nucleoplasm, Nucleus, SMN-Sm protein complex, Z disc
Calculated MW:
32kDa
Observed MW:
38kDa
This gene is part of a 500 kb inverted duplication on chromosome 5q13. This duplicated region contains at least four genes and repetitive elements which make it prone to rearrangements and deletions. The repetitiveness and complexity of the sequence have also caused difficulty in determining the organization of this genomic region. The telomeric and centromeric copies of this gene are nearly identical and encode the same protein. However, mutations in this gene, the telomeric copy, are associated with spinal muscular atrophy; mutations in the centromeric copy do not lead to disease. The centromeric copy may be a modifier of disease caused by mutation in the telomeric copy. The critical sequence difference between the two genes is a single nucleotide in exon 7, which is thought to be an exon splice enhancer. Note that the nine exons of both the telomeric and centromeric copies are designated historically as exon 1, 2a, 2b, and 3-8. It is thought that gene conversion events may involve the two genes, leading to varying copy numbers of each gene. The protein encoded by this gene localizes to both the cytoplasm and the nucleus. Within the nucleus, the protein localizes to subnuclear bodies called gems which are found near coiled bodies containing high concentrations of small ribonucleoproteins (snRNPs). This protein forms heteromeric complexes with proteins such as SIP1 and GEMIN4, and also interacts with several proteins known to be involved in the biogenesis of snRNPs, such as hnRNP U protein and the small nucleolar RNA binding protein. Multiple transcript variants encoding distinct isoforms have been described.
Purification Method:
Affinity purification
Gene ID:
6606
Clone Number:
ARC1710
Storage Buffer:
Store at -20℃. Avoid freeze / thaw cycles.Buffer: PBS with 0.02% sodium azide,0.05% BSA,50% glycerol,pH7.3.
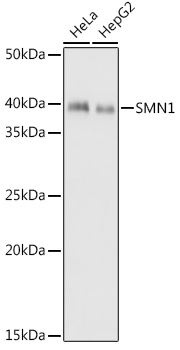
Western blot analysis of various lysates using SMN1 Rabbit mAb (CAB9707) at 1:1000 dilution.Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution.Lysates/proteins: 25μg per lane.Blocking buffer: 3% nonfat dry milk in TBST.Detection: ECL Basic Kit (AbGn00020).Exposure time: 1s.




")






