The ALG8 Polyclonal Antibody (PAC015386) is an essential tool for researchers studying the ALG8 protein, a key enzyme involved in the N-linked glycosylation pathway. This antibody, produced in rabbits, exhibits high reactivity specifically with human samples and has been validated for use in Western blot applications.ALG8 plays a vital role in the glycosylation process, which is essential for proper protein folding and function. Dysregulation of N-linked glycosylation has been linked to various diseases, including congenital disorders and cancer.
By targeting the ALG8 protein, researchers can investigate its role in disease pathogenesis and potentially develop new therapeutic strategies.With its ability to detect and analyze ALG8 in different cell types, this antibody is ideal for studies in molecular biology, biochemistry, and cell biology. By understanding the function and regulation of ALG8, researchers can gain valuable insights into disease mechanisms and potentially identify novel therapeutic targets.
Antibody Name:
ALG8 Antibody (PACO15386)
Antibody SKU:
PACO15386
Size:
50ul
Host Species:
Rabbit
Tested Applications:
ELISA, IHC
Recommended Dilutions:
ELISA:1:2000-1:5000, IHC:1:25-1:100
Species Reactivity:
Human, Mouse
Immunogen:
Fusion protein of human ALG8
Form:
Liquid
Storage Buffer:
-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
Purification Method:
Antigen affinity purification
Clonality:
Polyclonal
Isotype:
IgG
Conjugate:
Non-conjugated
The image on the left is immunohistochemistry of paraffin-embedded Human esophagus cancer tissue using PACO15386(ALG8 Antibody) at dilution 1/30, on the right is treated with fusion protein. (Original magnification: x200).
The image on the left is immunohistochemistry of paraffin-embedded Human thyroid cancer tissue using PACO15386(ALG8 Antibody) at dilution 1/30, on the right is treated with fusion protein. (Original magnification: x200).
Background:
This gene encodes a member of the ALG6/ALG8 glucosyltransferase family. The encoded protein catalyzes the addition of the second glucose residue to the lipid-linked oligosaccharide precursor for N-linked glycosylation of proteins. Mutations in this gene have been associated with congenital disorder of glycosylation type Ih (CDG-Ih). Alternatively spliced transcript variants encoding different isoforms have been identified.
Synonyms:
ALG8, alpha-1,3-glucosyltransferase
UniProt Protein Function:
ALG8: Adds the second glucose residue to the lipid-linked oligosaccharide precursor for N-linked glycosylation. Transfers glucose from dolichyl phosphate glucose (Dol-P-Glc) onto the lipid-linked oligosaccharide Glc(1)Man(9)GlcNAc(2)-PP-Dol. Defects in ALG8 are the cause of congenital disorder of glycosylation type 1H (CDG1H). CDGs are a family of severe inherited diseases caused by a defect in protein N- glycosylation. They are characterized by under-glycosylated serum proteins. These multisystem disorders present with a wide variety of clinical features, such as disorders of the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. Belongs to the ALG6/ALG8 glucosyltransferase family.Protein type: Transferase; Membrane protein, multi-pass; EC 2.4.1.265; Membrane protein, integral; Glycan Metabolism - N-glycan biosynthesisChromosomal Location of Human Ortholog: 11q14.1Cellular Component: endoplasmic reticulum membraneMolecular Function: alpha-1,3-mannosyltransferase activity; dolichyl pyrophosphate Man9GlcNAc2 alpha-1,3-glucosyltransferase activity; dolichyl-phosphate-glucose-glycolipid alpha-glucosyltransferase activityBiological Process: dolichol-linked oligosaccharide biosynthetic process; oligosaccharide-lipid intermediate assembly; protein amino acid N-linked glycosylationDisease: Congenital Disorder Of Glycosylation, Type Ih
UniProt Protein Details:
NCBI Summary:
This gene encodes a member of the ALG6/ALG8 glucosyltransferase family. The encoded protein catalyzes the addition of the second glucose residue to the lipid-linked oligosaccharide precursor for N-linked glycosylation of proteins. Mutations in this gene have been associated with congenital disorder of glycosylation type Ih (CDG-Ih). Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]





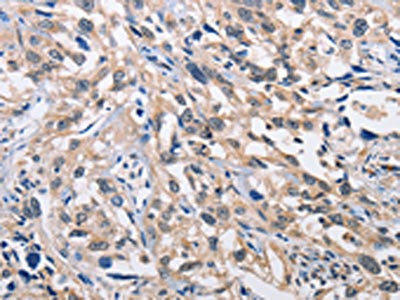
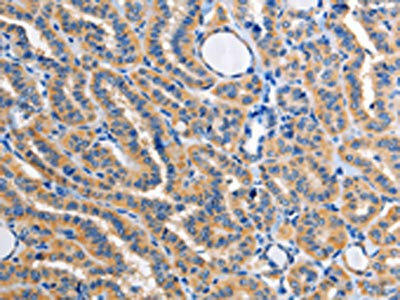

.")



.")
.")
. Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.")