The ACP2 Polyclonal Antibody (PACO18512) is a valuable tool for researchers studying ACP2, an enzyme that plays a crucial role in lysosomal function and protein degradation. This antibody, produced in rabbits, is highly specific to human samples and has been validated for use in Western blot applications. By binding to the ACP2 protein, this antibody allows for the detection and analysis of ACP2 in various cell types, making it an essential component of research in biochemistry and molecular biology.ACP2, also known as acid phosphatase 2, is involved in the breakdown of molecules within lysosomes, playing a vital role in cellular metabolism and nutrient recycling.
Dysregulation of ACP2 activity has been implicated in various diseases, including lysosomal storage disorders and certain types of cancer. By studying the function of ACP2, researchers can gain insights into these diseases and potentially develop targeted therapies to treat them.In summary, the ACP2 Polyclonal Antibody (PACO18512) is a reliable tool for investigating the role of ACP2 in cellular processes and disease pathogenesis. Its specificity and sensitivity make it an invaluable resource for scientists working in the fields of biochemistry, cell biology, and drug discovery.
Antibody Name:
ACP2 Antibody (PACO18512)
Antibody SKU:
PACO18512
Size:
50ul
Host Species:
Rabbit
Tested Applications:
ELISA, WB
Recommended Dilutions:
ELISA:1:1000-1:2000, WB:1:200-1:1000
Species Reactivity:
Human, Mouse, Rat
Immunogen:
Synthetic peptide of human ACP2
Form:
Liquid
Storage Buffer:
-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
Purification Method:
Antigen affinity purification
Clonality:
Polyclonal
Isotype:
IgG
Conjugate:
Non-conjugated
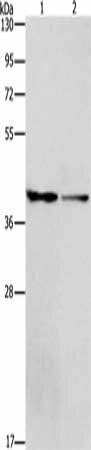
Gel: 8%SDS-PAGE, Lysate: 40 μg, Lane 1-2: 293T cells, hela cells, Primary antibody: PACO18512(ACP2 Antibody) at dilution 1/400, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 3 minutes.
Background:
This gene encodes the β subunit of lysosomal acid, phosphatase (LAP). LAP is chemically and genetically distinct from red cell acid, phosphatase. The encoded protein belongs to a family of distinct isoenzymes which hydrolyze orthophosphoric monoesters to alcohol and phosphate. Mutations in this gene or in the related α subunit gene cause acid, phosphatase deficiency. Multiple alternatively spliced transcript variants encoding different isoforms have been identified for this gene.
Synonyms:
acid, phosphatase 2, lysosomal
UniProt Protein Function:
ACP2: Defects in ACP2 are a cause of acid phosphatase deficiency (ACPHD). The clinical features are intermittent vomiting, hypotonia, lethargy, opisthotonos, terminal bleeding, and death in early infancy. Lysosomal acid phosphatase is deficient in cultured fibroblasts and multiple tissues. Belongs to the histidine acid phosphatase family.Protein type: Membrane protein, integral; Phosphatase (non-protein); Cofactor and Vitamin Metabolism - riboflavin; EC 3.1.3.2; Motility/polarity/chemotaxisChromosomal Location of Human Ortholog: 11p11.2|11p12-p11Cellular Component: lysosomal lumen; membrane; lysosome; lysosomal membrane; integral to membraneMolecular Function: acid phosphatase activityBiological Process: dephosphorylation; lysosome organization and biogenesis; skeletal developmentDisease: Acid Phosphatase Deficiency
UniProt Protein Details:
NCBI Summary:
This gene encodes the beta subunit of lysosomal acid phosphatase (LAP). LAP is chemically and genetically distinct from red cell acid phosphatase. The encoded protein belongs to a family of distinct isoenzymes which hydrolyze orthophosphoric monoesters to alcohol and phosphate. LAP-deficiencies in mice cause multiple defects including bone structure alterations, lysosomal storage defects in the kidneys and central nervous system, and an increased tendency towards seizures. An enzymatically-inactive allele of LAP in mice exhibited a more severe phenotype than the null allele, and defects included cerebellum abnormalities, growth retardation, hair-follicle abnormalities, and an ataxia-like phenotype. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Oct 2014]







.")
.")